







 Most of the information that we have about Pythagoras (c.570–c.495 BC) was written centuries after his death. It is difficult to know with any certainty which accounts are true.
Most of the information that we have about Pythagoras (c.570–c.495 BC) was written centuries after his death. It is difficult to know with any certainty which accounts are true.
He was born on the Greek island of Samos, in the eastern Aegean Sea, just off the coast of Turkey. The image at left is of the monument erected in his honour in the harbour at Pythagoreio. It depicts him completing a right-angled triangle.
His father, Mnesarchus, was a gem-engraver or a merchant who apparently travelled around the Mediterranean Sea by ship, trading and selling his wares. It is possible that his mother's name was Pythais (according to Apollonios of Tyana, a first century orator and writer).
As a young adult, he apparently travelled to Egypt and Greece. He probably studied with the priests (the learned class) in Egypt and possibly met the great philosopher, scientist, mathematician, and engineer, Thales of Miletus. According to the Syrian philosopher, Iamblichus (ca. 245-325 AD), Pythagoras studied in Egypt for 22 years and learned as much as he could, especially in astronomy and geometry. He was then taken captive by Cambyses II's soldiers who took him to Babylon where he studied arithmetic and music etc. with the Magi for a further 12 years before returning to Samos at about 56 years of age. If this is true, he may have been in Babylon during the last years of the Jewish exile there. Some accounts have him travelling as far as India in his search for knowledge. He is supposed to have returned to Samos in 520 BC (two years after Cambyses died) at around 56 years of age.
At about 530 BC, he moved to Croton, in the part of southern Italy known as Magna Graecia, where there were many Greek colonies. It was here that he quickly attained extensive influence and established some form of intellectual, religious, philosophical and political society based on an aesthetic lifestyle. To indicate some of the confusion about his life, Aristoxenus (b. c. 375, fl. 335 BC) claimed that Pythagoras was 40 years of age when he left Samos (and not 56 as Iamblichus claimed).
According to some accounts Pythagoras married a native of Croton named Theano, and had children (there is mention of a son, Telauges, and three daughters, Damo, Arignote, and Myia). Myia was supposed to have married Milo of Croton a follower of Pythagoras and reputedly the strongest historical Greek that lived in ancient times. I have written of Milo elsewhere (see my story of Chocolate and Mathematics).
Towards the end of Pythagoras' life, a significant conflict developed between Croton and another town, Sybaris, to the north west. Even though the citizens of Croton won the battle, further conflicts developed in the region, much of it against the Pythagoreans. One major attack was made on a group of Pythagoreans, possibly in Milo's house. The building was set on fire and many perished. One account has Milo rescuing Pythagoras from the burning building. The Pythagorean 'brotherhood' quickly ceased to exist.
Pythagoras left no texts but many mathematical and scientific discoveries have been attributed to him in the fields of mathematics, music, astronomy and medicine. More precisely, these discoveries have been attributed to the Pythagoreans, so it is not known how many could be personally ascribed to Pythagoras himself!
It has been claimed that he was the first man to call himself a philosopher (a lover of wisdom). Almost certainly, Pythagorean ideas greatly influenced Plato (428/427 or 424/423-348/347 BC) and through him, all of Western philosophy.
The Pythagorean Theorem
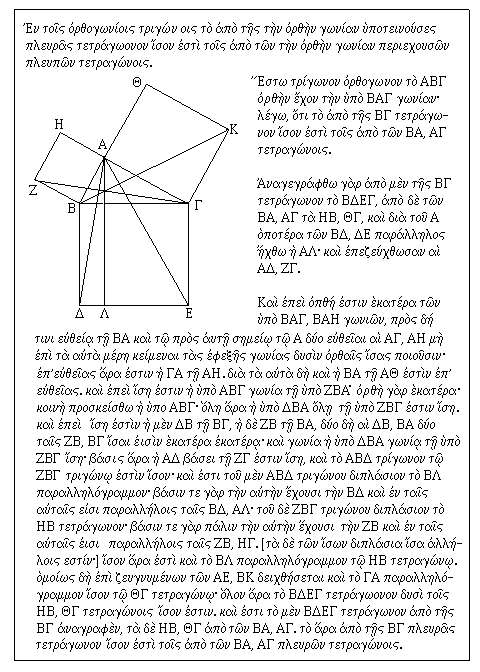
Although we know this theorem as Pythagoras' Theorem, the earliest writings connecting the theorem with his name date from five centuries after his death (in the writings of Cicero and Plutarch)! It was only in the fourth century AD, some 800 years after Pythagoras lived, that he was commonly given credit for discovering his theorem. It is said that he or his students were possibly the first to actually prove the theorem, although the Babylonians had very sophisticated knowledge of it (sufficient to suggest that they possibly had a proof of their own).
There is no doubt that the Pythagoreans regarded order in nature, and its links with number, with considerable awe. Plato's student, Aristotle, wrote in his Metaphysics 1–5 , cc. 350 BC, The so-called Pythagoreans, who were the first to take up mathematics, not only advanced this subject, but saturated with it, they fancied that the principles of mathematics were the principles of all things.
Music
It is thought that the Pythagoreans were the first to discover the harmonies of strings with lengths in particular ratios. In particular, it is said that they observed that the comparitive string lengths (when of the same material and tension) of the E, B and G strings of the cithara were in the ratio of 3:4:5.
Tetractys
 It has also been claimed that Pythagoras was fascinated by the tetractys, a triangular arrangement of ten objects as shown at right. Ten was considered a perfect number and, as a mystical symbol, the tetractys was very important to the Pythagoreans. Iamblichus reports that they would swear oaths by it.
It has also been claimed that Pythagoras was fascinated by the tetractys, a triangular arrangement of ten objects as shown at right. Ten was considered a perfect number and, as a mystical symbol, the tetractys was very important to the Pythagoreans. Iamblichus reports that they would swear oaths by it.
 Euclid (of Alexandria, fl.c. 300 BC) lived about 200 years after Pythagoras. He studied and taught in Alexandria at the time of
Euclid (of Alexandria, fl.c. 300 BC) lived about 200 years after Pythagoras. He studied and taught in Alexandria at the time of